Pavilion Publishing and Media Ltd
Blue Sky Offices Shoreham, 25 Cecil Pashley Way, Shoreham-by-Sea, West Sussex, BN43 5FF, UNITED KINGDOM
Tel: +44 (0)1273 434 943
Email: [email protected]


Motor neurone disease is a neurodegenerative disease, predominantly affecting the motor neurones. The number of people living with MND in the UK is 7,000 and 30,000 in the US, with an average age of onset at about 55 years. It is characterised by progressive weakness and wasting of limb, bulbar and respiratory muscles due to selective degeneration of both upper motor neurones and lower motor neurones.
Motor neurone disease (MND), also known as amyotrophic lateral sclerosis (ALS)/Lou Gehrig’s disease in the US and La Maladie de Charcot in France, is a rapidly progressive and ultimately fatal neurodegenerative disease. It was first described by a French neurologist Jean-Martin Charcot in 1886. It is the most common disorder of motor neurones and the third commonest neurodegenerative disease of later life (after Alzheimer’s disease and Parkinson’s disease).
MND is characterised by progressive weakness and wasting of limb, bulbar and respiratory muscles due to selective degeneration of both upper motor neurones (UMN) and lower motor neurones (LMN). The majority of cases are sporadic, affecting the elderly population in the sixth and seventh decades of life. However, approximately 5-10% of cases are familial, usually with autosomal dominant mode of inheritance, where an earlier age of onset is seen.
Mostly uniform all over the world, its annual incidence is two in 100,000 and a prevalence of approximately five per 100,000.1 There is a slight male predominance with a male to female ratio of 3:2. Patients with MND have an average life expectancy of 3–5 years, but survival of up to 10 years is seen in about 5% of patients. Despite this variability in prognosis, ALS invariably progresses to death and in the majority of cases this occurs as a result of respiratory muscle weakness and consequent respiratory failure. In the past 20 years, great strides have been made in understanding the pathophysiology of MND and managing its symptoms to sustain quality of life. MND, however, is currently incurable and no substantial disease modifying therapy is available.
Clinical manifestation of MND is wasting, weakness and spasticity of skeletal muscles that is usually focal in onset and gradually spreads to adjacent muscle groups. In the majority of cases the disease starts in upper or lower limbs and approximately 20% of patients initially experience bulbar, ie, speech or swallowing-related symptoms. Respiratory symptoms (eg. shortness of breath, orthopnoea) as a presenting feature occurs in about 2% of cases.
The characteristic signs of limb onset disease are asymmetrical distal muscle wasting and weakness, widespread fasciculations along with increased tone and brisk reflexes. This can result in walking difficulties and loss of hand dexterity. Bulbar symptoms include dysarthria, which often precedes dysphagia. Finally, features of respiratory involvement include dyspnoea, orthopnoea, a weak cough and also sleep related respiratory symptoms. A classical sign is “split hand,” ie, wasting of the thenar eminence and the first dorsal interosseous muscle while hypothenar eminence is spared.2
In the lower limbs, muscle wasting often involves tibialis anterior muscle, causing a foot drop (Figure 1). Tongue faciculations (tongue should be inspected inside the mouth) are highly specific for MND (Figure 2). It has also been recognised that in some patients non-motor symptoms may also develop such as pseudo bulbar affect and cognitive impairment; clinical frontotemporal dementia (behavioural variant) is observed in approximately 30% of MND patients. Pelvic floor muscles and extra-ocular muscles are typically spared and symptoms such as urinary incontinence and diplopia would suggest against the diagnosis of MND. Similarly, long finger flexor muscles and triceps remain strong till late and early involvement of these muscles should question the diagnosis. Absence of major sensory symptoms and signs is a pre-requisite for the diagnosis.
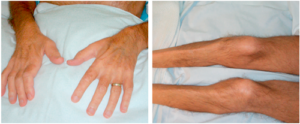
Figure 1: Wasting of the first dorsal interosseous and tibialis anterior in a patient with MND
As the disease progresses, limb, bulbar and respiratory muscles are progressively involved and become weaker, giving rise to increased symptoms and disability. Patients face profound physical impairment and reduced quality of life due to lack of ambulation, dysphagia, dysarthria and respiratory insufficiency.
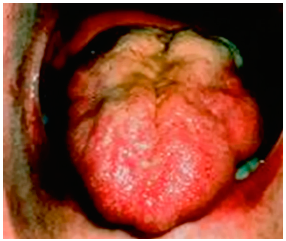
Figure 2: Bilateral tongue wasting in a patient with MND
Depending on the relative degeneration of upper or lower motor neurones, the disease can be divided into three sub-types, ie, primary lateral sclerosis PLS (clinically only UMN signs with no LMN signs developing for at least three years of follow-up), amyotrophic lateral sclerosis ALS (clinically mixture of UMN and LMN signs) (Figure 3) and progressive muscular atrophy PMA (clinically LMN signs only). ALS may be further sub-divided into UMN predominant or LMN predominant, depending on the predominant clinical features. ALS is considered as the classic form of MND as it is the most common sub-type (over 70% of cases) and both the PLS and PMA may transform into ALS phenotype after certain number of years. Thus, MND may be considered as a spectrum disorder with PLS and PMA at the two extremes. PLS and PMA carry better prognosis and in a patient with PLS or PMA, transformation into ALS phenotype may be considered as a marker of accelerated disease course.
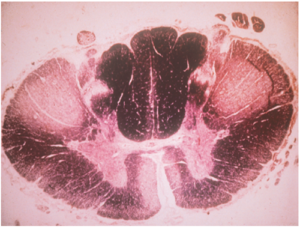
Figure 3: Spinal cord cross-section of a patient with MND. Note the pale anterior horns and antero-lateral corticospinal tracts
At least 22 different gene mutations have been identified in the families affected with MND (http://alsod.iop.kcl.ac.uk). The most common form of inheritance is autosomal dominant, although autosomal recessive and X-linked inheritance may also be seen in some families. The most recent discovery is the identification of intronic hexanucleotide expansion in the C9ORF72 gene (on chromosome 9p), which is responsible for up to 40% of familial cases and 7-8% of sporadic cases. The exact disease mechanism associated with C9ORF72 is unknown, abnormal C9ORF72 transcription and generation of toxic RNA foci have been postulated. Mutations in the SOD-1 gene (on chromosome 21q) encoding the free radical scavenging enzyme copper-zinc superoxide dismutase 1 may account for 20% of familial cases.
About 2-5% of familial ALS have mutations for TDP-43 gene (encoding TAR-DNA binding protein 43). These mutations are also rarely seen in apparently sporadic cases. Eight additional gene mutations (ASL2 – ALS8) have been identified as causative in rare cases of familial ALS. Many of them cause atypical, young onset and slowly progressive disease.
There is growing evidence that sporadic ALS also has a genetic basis. Several “susceptibility” gene mutations have been identified in sporadic cases (mostly using a candidate gene approach), which may act as genetic risk factors for developing the disease, in conjunction with other genetic and environmental factors. These include peripherin, apolipoprotein E and paraoxonase 1-3, to name a few.
MND is a clinical diagnosis. It is based upon the presence of a combination of both UMN and LMN signs and the exclusion of MND mimics following neurophysiological investigations and neuroimaging. The El Escorial criteria3 and its revised form are used to aid the classification of MND for research purposes and are also used to provide a level of diagnostic certainty. Patients can be classified as having either “Definite ALS”, “Probable ALS”, “Probable ALS – Laboratory Supported” or “Possible ALS”. A summary of the revised criteria is described in Table 1.
The diagnosis of ALS requires all of the following criteria:
Along with the absence of:
Definite ALS
Probable ALS
Probable ALS – Laboratory supported
AND
Possible ALS
OR
OR
The precise mechanism underlying selective cell death in motor neurone disease is currently unknown. Current understanding is that the loss of motor neurones is likely to be the end point of multiple pathogenic processes.4 A detailed discussion of these mechanisms is beyond the scope of this article; however, three most important mechanisms are listed below:
Neurones are large cells with high energy needs. There is high level of anti-oxidant enzyme, Cu/Zn superoxide dismutase 1, activity in motor neurones to combat with free radicals. Evidence of abnormal free radical metabolism in tissue obtained from patients with ALS and SOD 1 gene mutation linked to familial ALS supports the hypothesis of oxidative stress.5 Moreover, fibroblasts cultured from the skin of patients with ALS show more susceptibility to oxidative damage compared to fibroblasts from controls.
Mutations in two RNA binding proteins (TDP-43 and FUS) have been reported to cause MND. Aberrant mRNA splicing may have a critical role in the pathogenesis of ALS. Another two genes associated with FALS, senataxin and angiogenin also have a role in RNA metabolism.6
The neuronal cell bodies send information to the nerve terminals via a mechanism of axonal transport. This mechanism depends on a system of transport proteins (eg. dynactin, Kinesin) that may be dysfunctional in ALS. Axonal inclusions are seen in degenerating motor neurones, which represent abnormal assembly of axonal transport proteins. Mutations in the genes coding for the transport proteins are known to cause Charcot-Marie-Tooth disease. Mutation in microtubule associated protein tau gene is suspected in the pathophysiology of Guam variant of ALS.
A multidisciplinary approach to patient care lies at the heart of the management of MND and has been reported to significantly improve patient survival and quality of life.7 The management of MND will be discussed under the headings of life-prolonging measures, symptomatic therapies, occupational therapy and palliative care.
Since the 1990s several therapeutic compounds have been tested in clinical trials in patients with MND. Currently, riluzole (50mg BD) is the only disease modifying therapy available for ALS. Riluzole has several neuroprotective effects, but is predominantly a sodium channel blocker that inhibits the release of glutamate. The survival advantage provided by this neuroprotective agent was demonstrated in a randomised controlled trial, whereby treatment conferred a significant 35% reduction in the risk of death at 18 months.8 However, it only modestly prolongs lifespan, on average by three months, with some evidence that the survival benefit is transient. NICE has approved the use of riluzole for individuals with the ALS form of MND. Recently, FDA has approved a new medicine called Edaravone for use in patients with ALS.
Motor neurone disease linked to cholesterol imbalance in cells
Respiratory support interventions offer the largest survival and quality of life benefit currently available to the patients with MND.9 As a consequence of weakness in respiratory muscles, patients face two major consequences, ie, hypoventilatory respiratory failure and inability to cough effectively in order to clear airway secretions. Both of these problems can be addressed mechanically using non-invasive mechanical ventilator (NIV) and cough assist devices such as mechanical insufflator-exsufflator or lung volume recruitment bag (the breath-stacking technique).10
Due to important prognostic and therapeutic implications it is important that patients with MND are carefully screened for evidence of respiratory insufficiency during their follow-up. Clinical history of symptoms of respiratory failure is a helpful tool for the non-specialist. Symptoms such as orthopnoea, exertional dyspnoea, non-refreshing sleep, morning headaches and loss of appetite strongly suggest the presence of respiratory failure and need for respiratory support.
NIV has been demonstrated to improve symptoms such as resting dyspnoea, lack of energy, fatigue, day time hypersomnolence and also to improve survival by a median of seven months.11 Tracheotomy ventilation is generally not encouraged for patients with MND due to high costs, associated complications, unsustainable quality of life and terminal nature of the disease.
Patients with MND are prone to malnutrition due to dysphagia and a higher resting metabolic rate.12 Rapid weight loss is associated with a poor outcome13,14 and hence patients’ weight should be closely monitored.
Given the fact that MND is a hypermetabolic condition, diet rich in protein, polyunsaturated fatty acids and anti-oxidants (fruits, green tea) should be encouraged, although evidence to support these interventions is slim.15 Hypercholesterolaemia may be beneficial and statins should be avoided in patients with MND.16 Advice from a dietician and speech and language therapist is invaluable. In the initial stages dysphagia may be managed with changes in consistency of food and special swallowing techniques. Indications for gastrostomy include; losing 5% of weight from baseline, prolonged meal times, episodes of choking and aspiration. Role of gastrostomy should be discussed with the patient at an early stage, as poor respiratory function is associated with a high procedure-related mortality.17
Whilst it is accepted that enteral feeding via percutaneous endoscopic gastrostomy (PEG) improves patient nutritional status, a recent Cochrane Review highlighted the lack of robust data regarding its impact on survival.18 Whilst some studies have demonstrated PEG to provide no significant advantage,19 others have found it to prolong lifespan.20,21
There is evidence that good symptomatic care improves longevity and maintains quality of life.22
Pain
Pain in the limbs is a common complaint in advanced disease. Patients may have muscle pain (due to stiffness, spasticity or cramps), articular pain (due to immobility) or myofascial pain. Nature of the pain should be carefully assessed to select the appropriate remedy. Quinine sulphate is very effective for muscle cramps. Excessive muscle stiffness or spasticity may be helped with muscle relaxants (eg. baclofen, dantrolene, tizanidine). Dose should be carefully titrated as loss of muscle tone may worsen weakness. Pregabalin may be tried for myofascial pain. Physiotherapy, massage and regular re-positioning of the limbs should be combined with pharmacological measures.
Hypersialorrhea
Drooling of saliva may cause maceration of skin around the mouth and may be socially embarrassing. Production of saliva can be suppressed with anticholinergic drugs (eg. hyoscine transdermal patch, atropine eye drops administered sublingually) or botulimum toxin injection into the salivary glands. A portable suction device may be required in extreme cases. Good oral hygiene should be encouraged as suppression of saliva and poor oral hygiene may cause bad breath and oral thrush.
Thick sputum
Use of anticholinergics to control saliva may contribute to the crusting of sputum. Mucolytics such as carbocisteine or saline nebuliser are helpful to reduce the viscosity of sputum. Patients should be encouraged to maintain adequate hydration. Natural remedies such as papaya, pineapple juice can help.
Dyspnoea
Dyspnoea or orthopnoea may be a frightening symptom and may induce considerable anxiety. It usually indicates significant respiratory muscle weakness, but other causes such as a pulmonary embolism or chest infection should be considered. Non-invasive ventilatory support is effective in relieving dyspnoea. In the terminal stages of the disease oral morphine (2.5mg QDS) or sublingual lorazepam (1mg) may be required to control dyspnoea. Sublingual lorazepam may also be used for longer chocking episodes.
Urinary urgency
Detrusor over activity is a common symptom in patients with upper motor neuron disease. Antimuscarinic drugs such as oxybutynin, tolterodine or solifenacin are commonly used to reduce involuntary detrusor contractions. Anticholinergics should be used with caution in the elderly.
Constipation
Patients with MND are prone to constipation due to lack of activity, dietary consistency changes, use of anticholinergics and weak respiratory muscles (cannot generate sufficient intra-abdominal pressure). Oral laxatives may not be sufficient and measures like enema or manual evacuation may be required.
Fatigue/asthenia
Fatigue may be a direct symptom of the disease process. Patients should be carefully assessed for a secondary cause such as depression, nutritional insufficiency, respiratory failure or poor sleep.
Emotional lability
Anti-depressants like citalopram and amitriptyline are usually effective at controlling emotional lability. Nature of this symptom should be explained to the carer/spouse. Fluoxetine and sertraline have better side effect profile, if required for depression or anxiety and depression respectively.
Sleep disturbance
Cause of non-restorative sleep should be carefully ascertained. Common causes are anxiety/depression, orthopnoea or dyspnoea due to respiratory insufficiency, muscle cramps and uncomfortable posture due to inability to change position. Patients should be encouraged to sleep in an upright position.
Mobility aids
Simple orthotics such as foot-up splints may help improve mobility in the early stages of the disease. A community physiotherapist has an invaluable role in the continued assessment and provision of mobility aids eg, walking stick, Zimmer frame or wheelchair. Electronic wheelchair or mobility scooter may greatly enhance independence in patients with reasonable upper limb function. A head support especially for MND patients with head drop, due to neck muscles weakness, has recently been developed by a group of researchers at the University of Sheffield. Occupational therapists have essential role in assessing a patient at their residence and offer adaptive measures like stair lift, wet room or clos-o-mat shower toilets. Much can be done to improve the quality of life, but funding is often a limiting factor. Advice about funding should be available through the social worker.
Communication aids
Majority of the patients will eventually develop speech impairment. Modern communication devices are available for patients with dysphasia such as ipad, light writer or say-it-Sam tablet. Patients with no functional hand movement may use a head or foot mouse. In late stages, patients may be able to communicate with eye movements and picture charts only.
Patients should be linked into a palliative care team. There is substantial evidence that palliative care interventions improve quality of life for both patients and carers.23 Hospice referral is particularly important for patients with significant respiratory impairment who are intolerant to NIV. With good palliative care and support of family, the majority of deaths from MND can be peaceful. Opiates, benzodiazepines and oxygen are the most commonly used palliative care measures. Morphine, midazolam and glycopyrrolate may be given as a continuous infusion by syringe driver to maximise symptom control. Death generally occurs peacefully in sleep from hypercapnic coma.
In conclusion, in the past two decades MND has evolved from the therapeutic nihilism to a treatable condition. It is, however, still incurable. With improved (although still insufficient) understanding of its pathogenesis and genetics and the availability of high throughput drug screening technologies, research in stem cells and gene therapy, it is hoped that MND will soon enter in a new therapeutic era. MND sub-types due to single gene defects offer hope for interventions such as gene therapy and RNA silencing with antisense-oligonucleotide. Stem cell transplant to improve the internal milieu for motor neurones has also been shown to be a promising avenue.
Conflict of interest: none declared
Muhammad K Rafiq
Consultant Neurologist, Norfolk and Norwich University Hospitals NHS Trust
Motor neurone disease linked to cholesterol imbalance in cells

This website uses cookies to improve your experience. We'll assume you're ok with this, but you can opt-out if you wish. Accept Read more ...